Projects
The structure, function and evolution of coronavirus papain-like and 3C proteases and developing therapeutics targeted at these enzymes
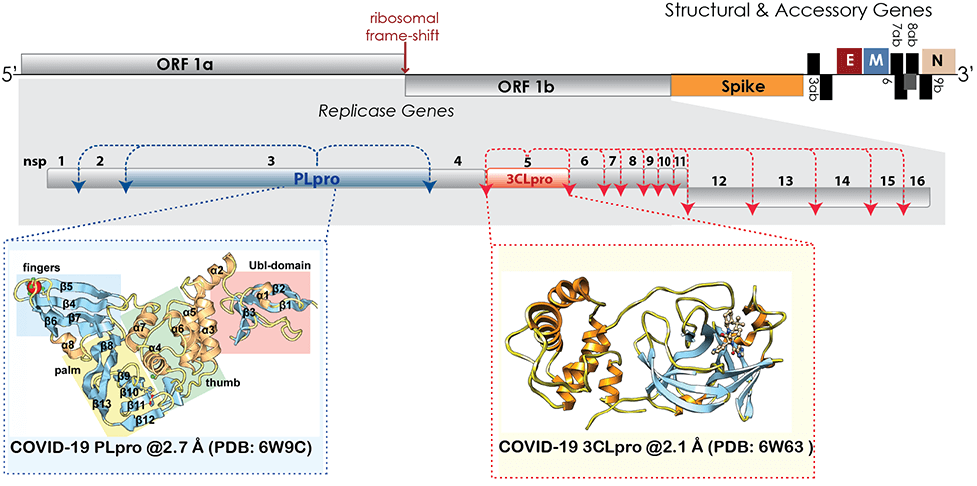
Coronaviruses are positive, single-stranded RNA viruses that encode a ~30kb genome. A crucial step for coronaviral replication is the processing of the ORF1 and ORF2 polyproteins to sixteen non-structural proteins (nsps) required for viral replication, RNA transcription and virus assembly. The CoV polyproteins are processed by two or three, depending on the CoV strain, viral proteases: the papain-like protease (PLpro) and 3-chymotrypsin like protease (3CLpro). These two enzymes are essential for viral replication and pathogenesis; therefore, they continue to be a target for developing therapeutics and antivirals.
PLpro is involved in processing the first three non-structural proteins but beyond its proteolytic activity, the Mesecar Lab discovered that CoVs have a “cloaking device” that helps them evade our innate immune response and the papain-like protease plays a major role in the cloaking mechanism by having additional deubiquitinating (DUB) and deISGylating activities. 3CLpro processes the other thirteen non-structural proteins housing proteins essential for genome replication and viral assembly.
The Mesecar Lab has been studying coronavirus proteases since the outbreak of severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002. Years of research from the Mesecar Lab along with others helped accelerate the development of antiviral therapeutics after the outbreak of severe acute respiratory syndrome-2 (SARS-CoV-2) in 2019 causing the worldwide pandemic of Coronavirus disease-2019 (COVID-19). In collaboration with Dr. Arun Ghosh in the Department of Chemistry at Purdue University and Dr. Richard Kuhn in the Department of Biological Sciences at Purdue University, the Mesecar Lab continues to discover and develop compounds that target CoV enzymes and then employs structure-based drug design to make potent, small-molecule therapeutics to treat CoV infections.
Understanding the mechanism, structure and function of newly discovered enzymes involved in inflammatory pathways in Alzheimer’s Disease (AD) and the discovery and development of small-molecule drugs to treat AD
Alzheimer’s Disease (AD) is currently the 6th leading cause of death in the United States. It is the most common form of age-related dementia, and it is estimated that 1 in 9 individuals over the age of 65 will develop AD. Despite its prevalence in the United States and across the globe, the only therapeutics available aim to manage symptoms of the disease and do not treat the underlying causes of AD.
Alzheimer’s Disease manifests itself in two major pathologies in the brain: the accumulation of amyloid plaques and the presence of neurofibrillary tangles. Much of the preliminary work in drug discovery focused on drugging the proteins responsible for the generation of these plaques and tangles. Despite decades of research from both academic and industry labs, there have been no FDA-approved therapeutics that successfully target these proteins. The Mesecar Lab studies one of the proteins directly involved in the generation of amyloid plaques called beta-secretase. Using structure-based drug design, we have been able to design potent small-molecule compounds that inhibit beta-secretase and, importantly, are selective for beta-secretase over other close homologs found in the body.
In addition to ongoing research aimed at stopping the production of plaques and tangles, the field has been exploring how inflammation in the brain also contributes to the progression of AD. The Mesecar Lab is fortunate to be a part of a national center aimed at identifying new and promising targets to treat AD. The center is a highly collaborative effort between Purdue, IU School of Medicine, and other similar centers around the United States. This center has put forward two new targets, a phosphatase and a phospholipase, that have been shown to have differential expression in AD brains as compared to healthy brains. Using structure-based drug design as well as a number of other biophysical techniques, the Mesecar Lab will explore substrate recognition, investigate oligomeric state, and probe ligand binding of these proteins. These new targets represent a promising new era of drug design efforts against AD.

The roles of deubiquitinating enzymes (DUBs) in cancer and antagonism of the innate immune response and targeting DUBs for anti-cancer and antiviral drug development
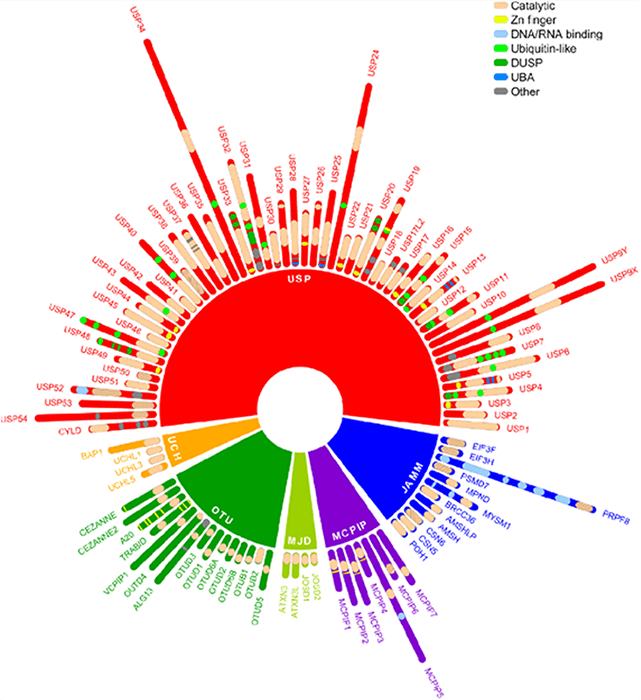
Ubiquitination is one of the most prevalent post-translational modifications involved in cellular pathways including cell-cycle progression, apoptosis, as well as gene regulation making it crucial for proper cellular function. Deubiquitinating enzymes (DUBs) are responsible for removing ubiquitin molecules from proteins that have been targeted for degradation, localization, or inactivation. The largest family of DUBS, ubiquitin-specific proteases (USPs), has at least 58 identified members, all of which contain a highly conserved catalytic domain. Due to their involved nature, particularly in cancers and neurodegenerative diseases, USPs have become promising leads for developing small molecule therapeutic inhibitors. Though many inhibitors have been identified for USPs, these compounds generally target the highly conserved active site and as a result have low specificity. In recent years, ancillary domains of USPs have become a focus in drug development because of their contribution to protein-protein interaction and substrate recognition.
Our lab has studied various DUB-enzymes including USPs, viral-DUBs, and other regulator proteins of the ubiquitination system in efforts of developing an anti-cancer therapeutic. Specifically, we have studied USP17, USP7, and Keap1 among others not noted in this summary. USP17 is a cytokine-inducible USP required for cell-cycle progression as it applies stress of the Ras converting enzyme 1 (RCE1) and Cell Division Cycle 25A (CDC25A) signaling pathways and is upregulated in breast and prostate cancer. USP7 is one of the most well-studied USPs and is involved in regulating the tumor suppressor p53/MDM2 pathway by silencing p53 through inappropriate stabilization of MDM2. This disruption of p53 signaling is responsible for about 50% of all human cancers and UPS7 has been upregulated in numerous forms of cancers and a promising target for anti-cancer therapeutics. Kelch-like ECH-associated protein 1 (Keap1) negatively regulates Nrf2, a transcription involved in activating the antioxidant response, whereby Keap1 facilitates the ubiquitination and proteolysis of Nrf2 leading to oxidative stress. There is an overexpression of Nrf2 in cancerous cells, used to promote cell growth and protect against oxidative stress and chemotherapeutics therefore, Keap1 is an attractive drug target.
In summary, we have enzymatically and biochemically characterized numerous USPs, viral-DUBs, and other regulator proteins of the ubiquitination system and have performed high-throughput drug screening on our most attractive leads along with structural analyses using both X-ray crystallography and cryo-electron microscopy.
Investigating the mechanism and structure of sulfotransferase enzymes to further understand their role in prostate cancer
Sulfotransferases (SULTs) are a family of phase II enzymes which catalyzes the sulfonation or the transfer of a sulfonate group (-SO3-1) from a universal donor, 3’-Phosphoadenosine-5’-phosphosulfate (PAPS), to a variety of hydroxyl (-OH) or primary amine (-NH2) containing substrates. Sulfonation is one of the major conjugation pathways responsible for the deactivation, detoxification and excretion of xenobiotic and endogenous molecules implicating SULTs and their regulations in the pathogenesis of many diseases.
Sulfotransferase 2B1b (SULT2B1b), an under-studied member of SULT family, is a 3β-hydroxysteroid sulfo-conjugating enzyme that exhibits abnormal expression levels in different types of cancers including prostate, breast, liver and colon. In the case of prostate cancer, SULT2B1b and cholesterol sulfate, the sulfonated product of cholesterol by SULT2B1b, have been identified as a potential drug target and a biomarker respectively, based on the observations of various in-cellulo studies. The major limitation in developing inhibitors for SULT2B1b is the lack of its proper biochemical characterization and the inconvenience of the available radiometric assay for routine and high throughput analysis. To address this gap in knowledge, our lab has developed SULT2B1b specific, continuous, fluorescent based, coupled-enzyme assay for the routine analysis of this enzyme. To validate our assay results we developed another assay based on desorption Electrospray Ionization Mass Spectrometry (DESI-MS) in collaboration with the Dr. Graham Cook’s Lab in the Department of Chemistry, Purdue University. The major goal of this collaborative project is the proper kinetic characterization of SULT2B1b and identifying small-molecule inhibitors using high throughput screening (HTS) studies. Latter part of the project involves characterization and validation of identified inhibitors using various biophysical and in-cellulo studies.
Sulfotransferase 1A1 (SULT1A1) is another member of SULT family that selectively catalyzes the sulfo-conjugation of phenolic compounds. This enzyme is one of the major enzymes in human liver which involves in detoxifying xenobiotic compounds. During the detoxification, it can bio activate dietary and environmental promutagens and procarcinogens. Several studies have demonstrated that dietary carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), induces prostate tumors in rats and sulfonation by SULT1A1 is the major route of metabolic activation of PhIP that can consequently exert mutagenic/carcinogenic effects by forming DNA adducts. Given these factors, it is plausible to consider SULT1A1 as a potential drug target in prostate cancer. Additionally, SULT1A1 is a major risk factor in liver, colorectal and breast cancers as well. Therefore, it is apparent the importance of identifying effective drugs targeting this enzyme.